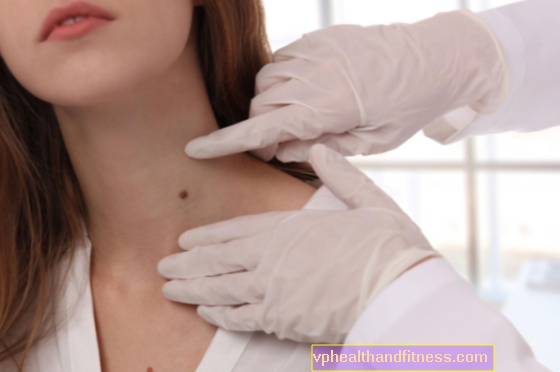
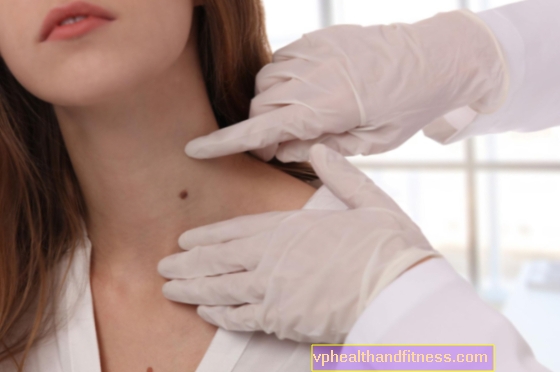
Phakomatosis, også kendt som hud- og nervesygdomme eller neuroectomesodermale dysplasier, er medfødte udviklingsforstyrrelser, der primært påvirker huden og nervesystemet. Abnormiteter påvirker ofte også andre organer, det er værd at finde ud af, hvem der er i fare for at udvikle sygdommen, og hvordan phacomatosis manifesterer sig.
Phakomatosis (fra græsk phakoma dvs. nevus, plet) er en gruppe på over 70 sygdomme eller rettere udviklingsforstyrrelser i forskellige væv, de mest almindelige er hudændringer og neurologiske symptomer, der indikerer ændringer i nervesystemet.
Disse abnormiteter er relateret til det faktum, at begge systemer har en fælles oprindelse - kimlaget kaldet ektoderm. Derudover kan skaden påvirke det vaskulære system, mere sjældent indre organer, og tendensen til neoplasma øges.
Phacomatosis: årsager og typer
Årsagen til phakomatosis er oftest mutationer af enkelte gener, der er karakteristiske for hver sygdom, deres virkning er en udviklingsforstyrrelse allerede i den 3-4. Uge i fostrets liv.
Selve navnet "phacomatosis" kommer fra det første udtryk for knuder på nethinden, der opstår i løbet af en af disse sygdomme - tuberøs sklerose.
Forekomsten af phacomatosis varierer meget, det anslås, at den mest almindelige forekommer hver 2000 fødsler, den sjældneste - hvert par millioner.
Risikoen for disse lidelser afhænger ikke af forældrenes livsstil eller graviditetsforløbet, de mutationer, der forårsager dem, vises ofte ved en tilfældighed. På grund af den genetiske årsag til disse sygdomme er der ingen effektiv terapi, og deres forløb er normalt kronisk og progressivt.
De mest almindelige phakomatoser er:
- neurofibromatose type 1
- neurofibromatose type 2
- tuberøs sklerose
- Sturge-Weber syndrom
- von Hippel-Lindau syndrom
- ataksi-telangiektasi
- Rendu-Osler-Weber sygdom
De sjældnere inkluderer:
- Farveinkontinens
- Gorlin-Goltz syndrom
- Schimmelpenning-Feuerstein-Mims syndrom
- Klippel-Trénaunay syndrom
Phacomatosis er en sjælden, men farlig genetisk sygdom, der er forbundet med hyppig kræft og ændringer i nervesystemet.
Meget mindre farligt, selvom forskellige hudlæsioner ofte er de første, der bemærkes, kan deres natur stille en ordentlig diagnose for lægen.
På trods af resultaterne inden for medicin og genetik er der ingen effektiv terapi til behandling af denne gruppe sygdomme, og patienter med sådanne diagnoser kræver hyppige kontroller af mange specialister, herunder en neurolog, øjenlæge, hudlæge, nefrolog, fysioterapeut og psykolog.
Håndteringen af sådanne patienter er baseret på behandling af læsioner, især neoplasmer, og hvis de forårsager ubehag, også hudlæsioner.
Type 1 neurofibromatose
Den mest almindelige phacomatosis, også kendt som von Recklinghausens sygdom, er autosomal dominerende arv, og forekommer derfor ofte i mange generationer af en familie, selvom det også er resultatet af en ny mutation.
Alle eller nogle af dem kan være til stede i neurofibromatose:
- hudændringer
- neurofibromer
- epilepsi
- knogledefekter
- nervesystemtumorer
Hudsymptomer er meget karakteristiske, findes hos alle patienter, de kaldes cafe au lait spots.
Disse fødselsmærker er lysebrune ændringer, ofte meget omfattende og forekommer overalt i kroppen, med alderen har de en tendens til at vokse, blive mørkere og virke nye.
Andre hudlæsioner er fregnede, fine pletter, der optræder i puberteten.
Neurofibromer er derimod røde eller blålige hævede bump, især på underlivet og ryggen, men kan også forekomme inde i kroppen.
Deres variation er plexus neurofibromer, som igen opstår som et resultat af fortykkelse af store nerver, hvorfor de oftest findes omkring øjenhulerne eller omkring halsen.
Væksten af sådanne neurofibromer kan være farlig, hvis de vokser ind i rygmarvskanalen, eller hvis den vokser inden i lemmerne og forårsager knogledeformation.
Blandt de knogledefekter, der oftest observeres i von Recklinghausen-sygdommen, er den laterale krumning af rygsøjlen (skoliose) og dens overdreven forreste krumning (kyphosis), der er også defekter i kraniet, der fører til de store hoved- og knogledefekter i underbenene.
Klumper på nethinden, et karakteristisk symptom på phakomatosis, i neurofibromatosis type 1 kaldes Lisch-knuder, er brune i farve og forekommer hos næsten alle patienter.
Von Recklinghausens sygdom er også forbundet med en større tendens til at udvikle neoplasmer i centralnervesystemet, de er hovedsageligt synsnerven gliomer, som kan forårsage synshandicap eller dysfunktion i hypofysen.
Diagnosen af type 1 neurofibromatose er baseret på det passende antal og størrelse af cafe au lait pletter, optisk nerve glioblastom, tilstedeværelsen af neurofibromer eller plexus neurofibromatose, Lisch knuder og tilstedeværelsen af patienter i familien.
Der er ingen effektiv kur, kun symptomatisk behandling er mulig med tidlig behandling af nervesystemets neoplasmer - radio- eller kemoterapi og hudlæsioner.
Derfor er hyppig sundhedsvurdering nødvendig: blodtryk, neurologisk kontrol, øjenundersøgelse og på grund af risikoen for CNS-neoplasmer, computertomografi eller resonansbilleddannelse hvert 2-3 år.
Den mest alvorlige sygdom, som er livstruende hos patienter med neurofibromatose type 1, er den hyppigere forekomst af ondartede svulster, herunder: tumorer i perifere nerver, sarkomer og leukæmi, hvorfor regelmæssig medicinsk kontrol er nødvendig.
Type 2 neurofibromatose
Det er omkring 50 gange mindre almindeligt end type 1 neurofibromatose, men ligesom det arves det på en autosomal dominerende måde, nogle symptomer er også ens, de inkluderer:
- pletter café au lait
- neurofibromer i huden
- tumorer i centralnervesystemet
Sidstnævnte er meget mere almindelige end i von Recklinghausen sygdom, og det mest karakteristiske er auditive neuromer, som forårsager høretab, ansigtsmuskelsvaghed, hovedpine og ubalance.
De symptomer, der kun forekommer i denne neurofibromatose, er uklarhed af linsen, dvs. grå stær, og beskadigelse af de perifere nerver.
Regelmæssig diagnosticering af patienter kræver hørevurdering (audiogram), hjerneafbildningstest, neurologiske og dermatologiske undersøgelser.
Der er ingen effektiv helbredelsesmetode, men overvågning og tidlig behandling af neoplastiske ændringer er vigtige.
Knoldsklerose
Knoldsklerose er en af de mest almindelige phakomatosis, manifesteret af hudlæsioner og udseendet af tumorer i nervesystemet, men også i øjnene eller indre organer, heldigvis er de ikke ondartede, menes det at være udviklingsmæssige abnormiteter.
Hudlæsioner, der er karakteristiske for denne sygdom, er de såkaldte farveløse modermærker - hvide pletter på huden på bagagerummet og lemmerne, i hvilket område der ikke er noget pigment.
Derudover er fibromer også karakteristiske, dvs. brune fremspring på panden og fingrene såvel som Pringles knuder, dvs. røde, små knuder placeret på næsen og kinderne.
Bortset fra hudlæsioner og tumorer har tuberøs sklerose også neurologiske sygdomme - epilepsi og mental retardation.
Diagnosen af sygdommen er ikke let, fordi sygdommen hos små børn normalt er let symptomatisk, og de diagnostiske kriterier inkluderer mere end 20 forskellige symptomer.
Tumorer i indre organer er fibroider (i hjertet) eller hæmangiomer og cyster (i nyrerne og leveren), de er ikke ondartede, men de kan skade organernes funktion - de forstyrrer blodgennemstrømningen eller forårsager nyresvigt.
Den eneste behandlingsmetode er symptomatisk behandling, dvs. tumorfjerning og farmakologisk behandling af neurologiske symptomer. Som ved anden phacomatosis skal sygdomsforløbet overvåges, og billeddannelsestest udføres periodisk.
Sturge-Weber syndrom
Dette er en meget sjælden phacomatosis. Karakteristisk for dette syndrom og nødvendigt for diagnose er en flad angioma i ansigtet, også kendt som en rødvinplet, dvs. en skarpt afgrænset rød, hævet plet.
Syndromet inkluderer også cerebrale hæmangiomer og epileptiske anfald, undertiden ledsaget af glaukom, parese og epilepsi. Billedbehandling kan vise kalkaflejringer i hjernen.
Behandling er symptomhåndtering, dvs. antiepileptika, glaukombehandling og kirurgisk fjernelse af hudlæsioner.
Von Hippel-Lindau syndrom
Dette syndrom er også en sjælden phakomatosis. I sin løb er den største bekymring den hyppige forekomst af tumorer, desværre mange steder på samme tid.
Organet, der er særlig udsat for det, er nyrerne, hvor både uskadelige nyrekyster og nyrekræft udvikler sig oftere.
Kræft påvirker nervesystemet og øjnene sjældnere, især lillehjernen, rygmarven og nethinden (hvor hæmangiomer udvikler sig her), symptomerne på disse tumorer er hovedpine, svimmelhed, balanceforstyrrelser, kvalme og opkastning og sensoriske forstyrrelser.
Fæokromocytom er en tumor i binyrerne, som også er hyppigere i von Hippel-Lindau syndrom end hos raske mennesker, og kan være ansvarlig for udviklingen af hypertension såvel som anfald af hovedpine, mavesmerter og hjertebanken.
Prædispositionen for kræft påvirker også mange andre organer, men de er ofte godartede, asymptomatiske cyster eller cystadenomer, organer, hvor bugspytkirtlen, det indre øre og epididymis hos mænd udvikler sig.
Håndtering af mennesker, der lider af von Hippel-Linadu syndrom, er hyppig profylakse, dvs. kontrol af organer, hvor kræft kan udvikle sig, dvs. regelmæssige besøg hos en øjenlæge, laboratorietest af urin og billeddannelse af underliv og hoved.
Ataksi-telangiektasi
I modsætning til de tidligere beskrevne arves phakomatoser autosomalt recessivt, så risikoen for at overføre sygdommen til børn af syge mennesker er meget lavere.
Symptomerne på denne sygdom er nedsat koordination (kaldet ataksi), udvidede, små blodkar synlige på huden, ofte kaldet edderkoppevene.
Derudover er personer med dette syndrom mere tilbøjelige til at lide af blodkræft og have immundefekt. Et karakteristisk træk ved denne sygdom er den høje følsomhed hos patienterne for ioniserende stråling, selv meget små doser forårsager deres død.
Rendu-Osler-Weber sygdom
Symptomerne og forløbet af denne sygdom er hovedsageligt forbundet med beskadigelse af væggene i blodkarrene. Derfor er telangiektasier og misdannelser, dvs. unormalt udvidede forbindelser af arterier med vener, sarte, hvilket resulterer i blødning fra næse, hud, lunger og særlig farlig blødning i centralnervesystemet.
Farveinkontinenssyndrom
Dette syndrom er en meget sjælden sygdom og er en af de få phakomatoser, hvis arv er relateret til køn.
Hudforandringerne indfases i naturen med blærer, vorter, misfarvning og endelig misfarvning og hårtab.
Læsionerne påvirker også øjnene og hjernen og forårsager blindhed og intellektuelt handicap.
---przyczyny-objawy-leczenie-rehabilitacja.jpg)


-u-15-rocznego-dziecka-porada-eksperta.jpg)